Hepcidin
Hepcidin Information
Hepcidin Structure
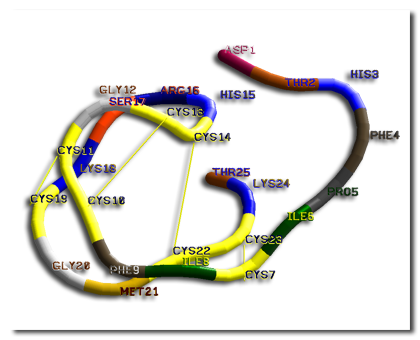
Hepcidin preprohormone, prohormone, and hormone size are 84, 60, and 25 amino acids, respectively. Twenty- and 22-amino acid forms of Hepcidin also exist in the urine. The N terminal region is required for function, and deletion of 5 N-terminal peptides results in a loss of function.
Hepcidin is also a tightly folded polypeptide containing 25 residues in length and is 32% beta sheets. The 25 amino acids have a hairpin structure and are stabilized by 4 disulfide bonds, which has been shown to act as the principal regulator of iron homeostasis in vertebrates. The structure of hepcidin has determined through the method of solution NMR. The different NMR studies showed a new model for hepcidin. Revealing at ambient temperatures, interconverts between two different conformations, which could be individually resolved by temperature variation. Using the different methods, the solution structure of hepcidin was determined at 325 and 253 K in supercooled water. The hepcidin conformation had appeared to be stabilized when the X-ray analysis of a co-crystal with Fab study was completed. This is similar to the high-temperature NMR structure.
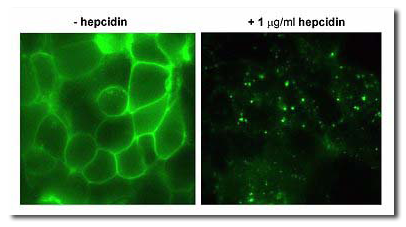
Hepcidin exerts its activity by binding to its receptor ferroportin. Ferroportin is a channel which exports iron from cells into plasma (panel A: cells expressing ferroportin-GFP fusion protein). When hepcidin binds to ferroportin, it causes ferroportin to be internalized and degraded (B). Internalized ferroportin cannot export iron out of the cells.
Hepcidin Function
The 25-amino acid peptide of hepcidin is secreted by the liver, which seems to be the “master regulator” of iron metabolism. Hepcidin inhibits iron transport by binding to the iron channel ferroportin, which is located on the basolateral surface of gut enterocytes and the plasma membrane of reticuloendothelial cells. Inhibiting ferroportin shuts off the iron transport out of these cells, which store iron. Ferroportin is also present on enterocytes and macrophages. By inhibiting ferroportin, hepcidin prevents enterocytes of the intestines from secreting iron into the hepatic portal system, thereby functionally reducing iron absorption. The iron release from macrophages is also prevented by ferroportin inhibition; therefore, the hepcidin maintains iron homeostasis. Hepcidin activity is also partially responsible for iron sequestration seen in anemia of chronic disease, and levels are elevated in people with renal failure.
Several mutations in hepcidin result in juvenile hemochromatosis. The majority of juvenile hemochromatosis cases are due to mutations in hemojuvelin, a regulator of hepcidin production.
Hepcidin has shown fairly consistent antifungal activity. Hepcidin’s antibacterial activity currently seems to be inconsistent. The current scientific evidence suggests that hepcidin is a central regulatory hormone, and its main action is to regulate systemic iron homeostasis.
Hepcidin History
The peptide was initially reported as LEAP-1, for Liver-Expressed Antimicrobial Protein and later became known as hepcidin.Independently, in a search for antimicrobial peptides, researchers working in the lab of Tomas Ganz discovered a peptide associated with inflammation, and named it “hepcidin” after observing that it was produced in the liver (“hep-“) and appeared to have bactericidal properties (“-cide” for “killing”). Both groups were focused on the antimicrobial properties of the peptide.
Hepcidin was first discovered in human urine and serum in 2000. Most understandings of hepcidin regulation and action comes from in vitro and mice studies that often use hepcidin mRNA expression as a read-out. Carrying out studies in humans is difficult due to the lack of suitable hepcidin assay. With the recent developments of assays to measure hepcidin in serum and urine, new opportunities to study the regulation of hepcidin in humans have arisen. Only a few laboratories are able to perform these assays at the current moment. The aim of the studies is to discuss insights into hepcidin regulation obtained from recent clinical studies in the light of findings from in vitro and mice studies. Ongoing studies in humans should provide us with more information on the etiology of iron metabolism disorders in order to create new therapeutic strategies and improve differential diagnosis protocols for these diseases.
Soon after this discovery, researchers discovered that hepcidin production in mice increases in conditions of iron overload as well as in inflammation. Genetically modified mice engineered to overexpress hepcidin died shortly after birth with severe iron deficiency, again suggesting a central and not redundant role in iron regulation. The first evidence that linked hepcidin to the clinical condition known as the anemia of inflammation came from the lab of Nancy Andrews in Boston when researchers looked at tissue from two patients with liver tumors with a severe microcytic anemia that did not respond to iron supplementation. The tumor tissue appeared to be overproducing hepcidin, and contained large quantities of hepcidin mRNA. Removing the tumors surgically cured the anemia.
Taken together, these discoveries suggested that hepcidin regulates the release of iron in the body.
Hepcidin’s Clinical Significance
Beta-thalassemia is one of the most common congenital anemias arising from partial or complete lack of β-globin synthesis. Excessive iron absorption is one of the main features of β-thalassemia and can lead to severe morbidity and mortality. The serial analyses of β-thalassemic mice indicate hemoglobin levels decreases over time, while the concentration of iron in the liver, spleen, and kidneys markedly increases. The overload of iron is associated with low levels of hepcidin. It was found that patients with β-thalassemia also have low hepcidin levels. The observations led researchers to hypothesize that more iron is absorbed in β-thalassemia than is required for erythropoiesis and whether the concentration of hepcidin is increasing in the body of such patients might be therapeutic, limiting iron overload. It was demonstrated that a moderate increase in expression of hepcidin in β-thalassemic mice limits iron overload, decreases formation of insoluble membrane-bound globins and reactive oxygen species, and improves anemia. Mice with increased hepcidin expression also demonstrated an increase in the lifespan of their red cells, reversal of ineffective erythropoiesis and splenomegaly, and an increase in total hemoglobin levels. The data led the researchers to suggest therapeutics that could increase hepcidin levels or act as hepcidin agonists might help treat the abnormal iron absorption in individuals with β-thalassemia and related disorders.
Featured Hepcidin Publications:
– Hepcidin, a Urinary Antimicrobial Peptide Synthesized in the Liver*
– Immunoassay for human serum hepcidin
– Hepcidin and iron regulation, 10 years later
Iron is essential for every living organism. It is a key component of blood hemoglobin, energy-generating enzymes, and other building blocks of life. The recently discovered hormone, hepcidin, controls the absorption and distribution of iron in the body.Hepcidin diagnostics and hepcidin-targeting drugs will revolutionize the medical management of anemias and iron overload diseases.
Prevalence of Iron Diseases. The prevalence of all types of anemia is staggering with ~1 billion humans worldwide affected at any time. Additionally, hereditary hemochromatosis (genetic iron overload disease) occurs in 10M patients worldwide. Iron-loading anemias such as beta-thalassemia affect another 15M patients. Hepcidin concentrations are altered in all these disorders and are expected to provide useful diagnostic and prognostic information.
Current diagnostic tests for iron status (serum ferritin, serum iron, transferrin saturation) do not provide adequate information in many diseases. Key markets for our hepcidin IVDs may include patients with anemia where hepcidin can be used to differentiate between iron deficiency anemias (IDA) and anemia of inflammation (AI). In hereditary hemochromatosis, hepcidin can differentiate those patients that need lifelong treatments from others whose disease is so mild that it does not require treatment.
In iron-loading anemias, hepcidin will be a prognostic tool to gauge the severity of the disease and its complications, and to assess the effectiveness of therapies. Iron deficiency and inflammation contribute to cancer- and chemotherapy-induced anemia (CIA; ~6- 8M WW, 60% anemic) and chronic kidney disease anemia (CKD; pre-dialysis 2M WW: 40% anemic; dialysis 700,000 WW: 90% anemic). ILS hepcidin IVDs could be used routinely to monitor iron status in CIAs and CKD patients over many years, and assess the effectiveness of EPO, anti-inflammatory, and dialysis therapies.
Other Hepcidin Background Information
Hereditary hemochromatosis: genetic complexity and new diagnostic approaches.
Since the discovery of the hemochromatosis gene (HFE) in 1996, several novel gene defects have been detected, explaining the mechanism and diversity of iron-overload diseases. At least 4 main types of hereditary hemochromatosis (HH) have been identified. Surprisingly, genes involved in HH encode for proteins that all affect pathways centered around liver hepcidin synthesis and its interaction with ferroportin, an iron exporter in enterocytes and macrophages. Hepcidin concentrations in urine negatively correlate with the severity of HH. Cytokine-mediated increases in hepcidin appear to be an important causative factor in anemia of inflammation, which is characterized by sequestration of iron in the macrophage system. For clinicians, the challenge is now to diagnose HH before irreversible damage develops and, at the same time, to distinguish progressive iron overload from increasingly common diseases with only moderately increased body iron stores, such as the metabolic syndrome. Understanding the molecular regulation of iron homeostasis may be helpful in designing innovative and reliable DNA and protein tests for diagnosis. Subsequently, evidence-based diagnostic strategies must be developed, using both conventional and innovative laboratory tests, to differentiate between the various causes of distortions of iron metabolism. This review describes new insights in mechanisms of iron overload, which are needed to understand new developments in diagnostic medicine.
PMID: 16627556 [PubMed – indexed for MEDLINE]
Iron homeostasis: fitting the puzzle pieces together.
Here, recent insights into iron homeostasis are highlighted. Three studies demonstrate the importance of the IRE-IRP system for enterocytes in balancing extracellular iron demand against cellular iron requirements, show that the hemochromatosis protein HFE exerts its iron-regulatory activity principally in hepatocytes by modulating the production of hepcidin, and provide strong support for a proposed mechanism of transcriptional regulation of hepcidin through a signaling cascade initiated by holotransferrin displacing HFE from transferrin receptor 1.
PMID: 18396134 [PubMed – indexed for MEDLINE]
Hepcidin: from discovery to differential diagnosis.
Although iron is essential for living organisms to survive, its reactive properties require strict regulation in order to prevent toxic effects. Hepcidin, a liver produced peptide hormone, is thought to be the central regulator of body iron metabolism. Its production is mainly controlled by the erythropoietic activity of the bone-marrow, the amount of circulating and stored body iron, and inflammation. Recent reports, however, provide new hypotheses on how hepcidin might exert its regulatory function. Although hepcidin was first discovered in human urine and serum, most of our understanding of hepcidin regulation and action comes from in vitro and mice studies that often use hepcidin mRNA expression as a read out. The difficulties in carrying out studies in humans have mostly been due to the lack of suitable hepcidin assay. The recent development of assays to measure hepcidin in serum and urine has offered new opportunities to study hepcidin regulation in humans. However, for the moment, only a small number of laboratories are able to perform these assays. The aim of this review is to discuss insights into hepcidin regulation obtained from recent clinical studies in the light of findings from in vitro and mice studies. Ongoing studies in humans should provide us with more information on the etiology of iron metabolism disorders in order to create new therapeutic strategies and improve differential diagnosis protocols for these diseases.
PMID: 18166790 [PubMed – indexed for MEDLINE]
Hereditary haemochromatosis: novel genes, novel diseases and hepcidin.
Since the discovery of the HFE gene of hereditary haemochromatosis in 1996 several new genetic defects have been identified, enabling explanation of the cause and variety of this disease. To date, at least 5 major types of hereditary haemochromatosis have been recognised. All these genes encode for proteins that are involved in metabolic pathways relevant to hepcidin synthesis in the liver. Hepcidin is a small protein that regulates the activity of the iron exporting protein ferroportin in the basolateral membrane of duodenal cells and the cell membrane of macrophages and thereby controls serum iron concentration. Plasma hepcidin concentration is elevated in body iron excess and by inflammatory stimuli, and is lowered in erythroid iron demand, hypoxia and most types of hereditary haemochromatosis. It is the clinician’s task to diagnose hereditary haemochromatosis before irreversible tissue damage arises and at the same time to differentiate between ongoing iron accumulation and increasingly prevalent disorders with elevated serum ferritin such as the metabolic syndrome.
PMID: 17557668 [PubMed – indexed for MEDLINE]